Very Long Chain Acyl CoA Dehydrogenase Deficiency (VLCADD) OMIM #201475
Edited by: Frances Rohr, MS RD LD, Children's Hospital, Boston, MA and Sandy van Calcar, MS RD, University of Wisconsin, Madison, WI
Last Updated: September 4, 2008
Background
Definition: Very Long Chain Acyl CoA Dehydrogenase Deficiency (VLCADD) is an autosomal recessive disorder caused by mutations in the acyl-coenzyme A dehydrogenase gene leading to insufficient enzymatic activity to allow complete mitochondrial beta-oxidation of long chain fatty acids. Long chain fats contain carbon lengths of 14 or greater. Further description of VLCADD
Incidence: The exact incidence of VLCADD is not established. It is estimated to affect 1 in 40,000 to 120,000 people.
Pre-symptomatic detection: It is possible to detect VLCADD through newborn screening with tandem mass spectrometry. The analyte that is elevated on newborn screening is the C14:1 acylcarnitine. An initial concentration of C14:1 over 1 umol/L is indicative of VLCADD, whereas C14:1 < 1 umol/L may indicate carrier status or milder phenotypes (1). The ratio of C14:1 to C12:1 acylcarnitines is usually elevated. Other long chain acylcarnitines may also be elevated. Each state newborn screening program sets specific criteria for a positive screen value.
Genetics: VLCADD is inherited in an autosomal recessive manner. A genotype:phenotype correlation is found, with the relatively common missense mutations, T220M and V243A, usually being associated with a milder form of the disorder and the null mutation R249W leading to a more severe form (2).
Confirmatory testing: Individuals with a positive newborn screening are seen at metabolic centers for further testing before the diagnosis can be confirmed. Confirmatory testing usually includes plasma acylcarnitine profile, plasma carnitine, urine organic acids, enzyme analysis, and mutational analysis. See American College of Medical Genetics Guidelines.
Signs and Symptoms
The severity of symptoms varies from the severe form (cardiomyopathy, non-ketotic hypoglycemia, death if untreated) which usually presents in infancy, to milder forms (non-ketotic hypoglycemia and hepatic dysfunction) to later onset VLCADD (muscle fatigue and rhabdomyolysis, usually in conjunction with prolonged exercise) (3). There is a broad clinical spectrum of VLCADD and individuals may exhibit some or all of these symptoms at various ages (4).
Laboratory Findings: Laboratory results should be compared to the reference ranges provided by the testing laboratory. Results will vary depending on the individual’s age, severity of disease, treatment modalities, and state of health. Not all findings are always present, even during acute episodes.
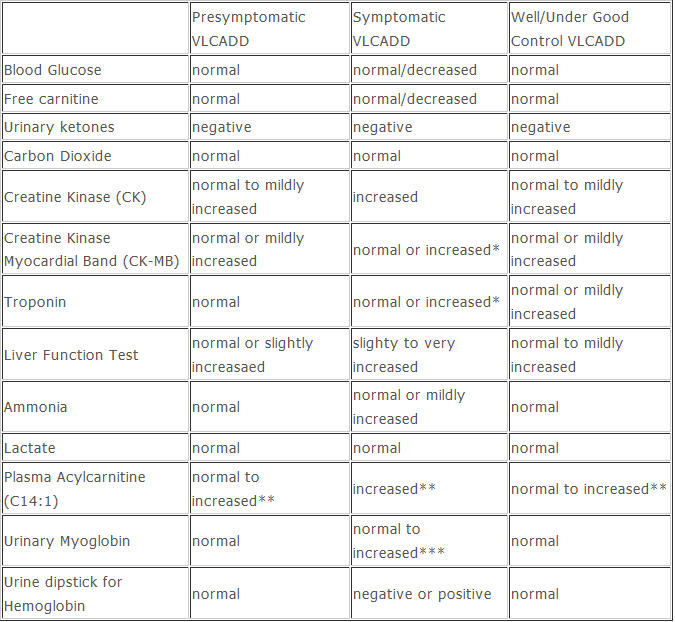
*presence of cardiac markers depends on specific mutations which cause cardiac involvement
**additional long chain acyl carnitine species may also be present
** *urinary myoglobin will clear much more quickly than ≠CK in blood
Biochemical Basis of the Disorder
Oxidation of Fats: When fat is needed as an energy source during periods of decreased intake, prolonged fasting or increased energy demands due to fever or stress, fatty acids are liberalized from adipose tissue, and activated to fatty acid acyl CoA. The fatty acid acyl CoA requires carnitine for transport into the inner mitochondria. (Note that medium chain (C8-C12) and short chain (C4-C6) fatty acids do not require carnitine for transport into the mitochondria). The long chain fatty acid acyl CoA is then split from the carnitine and is oxidized to acetyl CoA, which feeds into the Kreb’s cycle or is used for ketogenesis. A series of four enzymes are needed for each two-carbon unit that is cleaved from the fatty acid chain in the oxidation process. The enzyme used depends on the carbon chain length and include a dehydrogenase (VLCAD, MCAD or SCAD), a hydratase, a second dehydrogenase (LCHAD, SCHAD,) and a thiolase (5)
Individuals with VLCADD often exhibit episodes of non-ketotic hypoglycemia with vomiting and/or lethargy. This happens because ketones can not be produced when metabolism of fat is incomplete. Fatty acid metabolites accumulate and result in the inhibition of gluconeogenesis and abnormal liver function. Tissues that are highly energy dependent, such as cardiac and skeletal muscle, are deprived of energy and become impaired, causing cardiomyopathy and rhabdomyolysis.
Chronic Management: Experience treating infants with VLCADD is limited and treatment practices vary. These guidelines are based on review of the literature, abstracts presented at professional meetings, personal communication, and practice guidelines of several metabolic centers (6). GMDI expects that with more experience the guidelines will be revised and invites you to add comments about your experience by clicking on the “Comments” section at the end of the protocol.
Goals: The primary goal of diet treatment is to limit dependence on long chain fat as a substrate for energy production by preventing catabolism and limiting the amount of dietary long chain fat while providing adequate nutrients for normal growth and development. Acute illness is often the cause of metabolic decompensation due to poor intake and increased energy demands. Families should be provided with an emergency protocol and advised to seek medical treatment if the child has an acute illness accompanied by poor feeding, vomiting or lethargy.
Frequent feedings: Frequency of feeding depends on the severity of the disorder and the age and size of the child. A general guideline for infants is that they should be fed at least every 4 hours for children age 0-4 months, and one additional hour per month of age thereafter (e.g. 5 hours for 5 months, 6 hours for 6 months) up to 8 hours. In older infants, children, and adults, regular meals and snacks during the day and before bed are important to prevent hypoglycemia, fatigue or lethargy. Cornstarch therapy may be needed to prevent overnight hypoglycemia. Additional caloric support may be needed before and during exercise (see special circumstances).
Diet composition in infancy: Total fat intake from all fat sources (medium chain triglycerides (MCT), long chain fat and supplements) provides 40-45% of energy intake in infancy. In severe VLCADD characterized by cardiomyopathy and liver disease, long chain fat is restricted to 10 % and MCT to 30% of energy intake (7). For infants with moderate or mild forms of VLCADD, half of the fat calories are provided from MCT and half from long chain fat. An alternative method for determining the amount of MCT is provide 2-3 g MCT per kg body weight (5). Special formulas containing MCT are available for treatment of long chain fatty acid oxidation disorders. Breast milk or standard infant formulas may be used as the source of long chain fat. Protein should supply about 15% of energy intake.
The source of long chain fat may also be important. A diet with a higher percentage of essential fatty acids, linoleic and linolenic acid, result in less accumulation of disease-specific acylcarnitines than diets that contain a higher percentage of palmitic and oleic acid (8).
When an infant comes to attention through newborn screening, it may take several weeks to further define the severity of the disorder, and even then it is not known when or if the infant will become symptomatic, leaving clinicians uncertain regarding how aggressively to treat. One approach is to treat all infants with the strictest diet (10% of energy as long chain fat) and then liberalize fat restriction in those who are identified as having milder forms. Another is to treat only those infants who are symptomatic with the severe fat restriction and provide about 20-25% of energy as long chain fat in infants who are asymptomatic. In all cases, fasting avoidance and vigilance during acute illness is crucial.
Diet composition in children and adults: Diet composition in children and adults: The amount of total fat needed in the diet is reduced to about 35% of energy after approximately one year of age. 10-20% of total energy intake is provided by long chain fat and the remainder is supplied as MCT. This is approximately equivalent to 1-1.5 g MCT/kg/day. MCT may be supplied through special formulas or added as MCT oil to skim milk or other foods. Some centers use a modular protein supplement and add the required amount of MCT to it. As solid foods become more important in the diet, they supply the majority of long chain fat. Attention must be given to the sources of fat to ensure adequate essential fatty acids in the diet.
Monitoring
Essential Fatty Acids: The requirement for the essential fatty acids, linoleic and linolenic acid, should be met. Linoleic acid (C18:2n6) should comprise 3% of energy intake and linolenic acid (C18:3n3) should comprise 1% of energy intake. The DRI for essential fatty acids is higher than the FAO recommendations of 600 mg of linoleic acid and 50 mg of alpha-linolenic acid per kilogram body weight in term infants. The recommended ratio of linoleic to linolenic acid is between 5:1 and 10:1. Supplementation with specific oils, such as canola, walnut, or flaxseed oil may be necessary to meet essential fatty acid requirements. (See Sources of Essential fatty Acids)
Docosohexanoic acid (DHA): DHA can be derived from dietary omega-3 fatty acids, but since these are limited in the diet, some centers recommend supplementation. The dose (used in LCHAD, another long chain fatty disorder) for children weighing < 20 kg is 65 mg/day and for individuals weighing > 20 kg is 130 mg/day (9).
L-Carnitine: Plasma free carnitine is often low in VLCADD as fatty acids conjugate with carnitine and are excreted as acylcarnitine. Supplementation is recommended by some centers.The dose is 50-100 mg/kg, beginning with the lower dose in infancy.
Triheptanoin (C7): This odd chain triglyceride has been used experimentally in VLCADD patients to enhance availability of propionyl CoA as a citric acid cycle intermediate and thereby restore energy production (10).
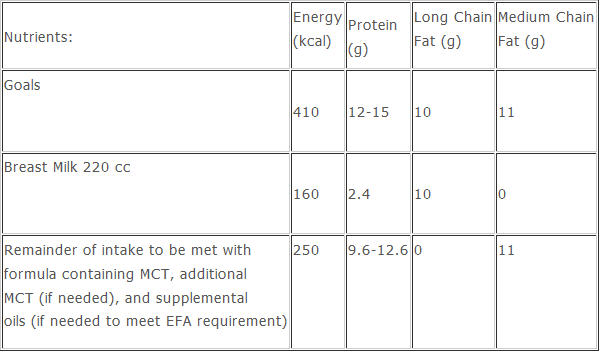
Sample diet calculation for 2 week old, 3.7 kg male with VLCADD (identified through newborn screening and asymptomatic)
Energy requirement = 410 kcal
45% of energy as fat = 185 kcal
50% of fat calories as long chain fat (9 kcal/g) = 92 kcal= 10 g
Breast milk per 100 cc contains 4.6 g fat = 220 cc breast milk
50 % of fat calories as MCT (8 kcal/g) = 92 kcal =11 g
Protein- 12-15% of kcal
Supplemental EFA, DHA, and L-carnitine as needed:
Anthropometrics: Normal growth is expected in VLCADD. Be mindful that children with fatty acid oxidation defects should not become overweight as weight reduction mobilizes fat stores. Caretakers should be advised that frequent feeding does not mean over-feeding.
Monitoring: Plasma acylcarnitine profile: Acylcarnitines are measured at each clinic visit. The goal is to maintain normal values of C14 and higher acylcarnitines, although long chain acylcarnitine profiles may not completely normalize with dietary treatment in some children. Acylcarnitines C8-C12 may be higher than normal due to MCT therapy.
Erythrocyte free fatty acid profile: Monitor at least twice yearly. Erythrocyte free fatty acids give a longer term view of fat intake than plasma concentrations. The essential fatty acids C18:2(n-6) (linoleic acid) and C18:3(n-3) (linolenic acid) are of particular importance.
Triene:tetraene ratio: Ratio should be <0.4. Trienes accumulate in essential fatty acid deficiency. If the ratio of trienes:tetraenes is elevated, re-evaluate essential fatty acid intake. This ratio is particularly sensitive to deficiency of linoleic acid and other n-6 fatty acids.
Arachidonic acid/DHA ratio of 3.4 ± 1.3 can also be useful to detect essential fatty acid deficiency, as long as the plasma concentrations of both fatty acids are adequate.
Plasma free carnitine is maintained in the normal range. Total and esterified carnitine levels may be high, especially if patient is on supplemental L-carnitine.
Urine organic acids- normal pattern is expected; dicarboxylic acids should not be present in healthy children with VLCADD.
Urine dipstick for myoglobinuria is recommended by some centers as a means to detect rhabdomyolysis.
Dietary Assessment: Three day food intake records or diet recall should be evaluated with particular attention to: Energy intake- inadequate energy intake will cause catabolism of fat stores and excessive energy intake increases the risk of obesity.
Diet composition: Type and amount of fat as prescribed; adequacy of essential fatty acids
Vitamins and minerals should meet DRI. Fat soluble vitamin intake may be inadequate, especially if a medical food is not being used.
back to top
Acute Management
At home care during illness:
Avoidance of fasting: Make certain that the child is meeting fluid and glucose requirements. Ensure that the child is still receiving carnitine and MCT oil.
Monitoring during illness: Signs and symptoms to watch for include muscle aches and pain, unusual fatigue, and dark urine color (or urine positive for myoglobinuria). Over time, caregivers will begin to recognize when an illness can be managed at home and when more serious intervention is required. As a general guideline, if an illness continues after 3 days of home treatment, it is recommended that caretakers contact their healthcare professional even if they do not feel that the illness is serious. Health care providers should respect and listen to the opinion of the people who know the child best.
Emergency Care: The responsibility for emergency management lies with different professionals in different institutions. These are the principles that need to be considered:
An emergency protocol should be followed whenever possible. Emergency care includes:
Glucose Administration: Caregivers can provide Instaglucose or cake icing gel to the child on the way to the hospital. Once in the emergency room IV Glucose (D10) is given at 1.5 times maintenance.
IV carnitine may also be necessary.
MCT should be provided as soon as the child is able to tolerate it by mouth or by tube-feeding, since no IV solution of MCT is available.
Intralipid is contraindicated
Special Circumstances
Exercise increases energy demands. Children with VLCADD can participate in physical education or sports activities, but should have a snack before exercise and during prolonged exercise. In order to prevent muscle from using fat as an energy substrate, a constant supply of glucose is necessary. Complex carbohydrates such as low fat granola bars, low fat whole grain bread or cereal products, or cornstarch are the best choice for snacks before and during exercise. Provision of MCT in snacks may also improve exercise tolerance and reduce rhabdomyolysis (11). If fatigue or muscle pain ensues, rest, extra fluid and carbohydrate-containing snacks are encouraged. If muscle pains are severe and do not improve after rest, a physician should be contacted
Catabolic stress may also be caused by surgery, trauma, labor, childbirth and the post-partum period. Provision of IV caloric support and MCT supplementation is needed.
Education
Learning Objectives: The individual with VLCADD and/or the caregiver will:
- Understand importance of avoiding fasting and eating meals on a regular schedule
- Understand the emergency protocol and when to use it
- Recognize signs of illness
- Offer foods/formulas with appropriate type and amount of fat
- Recognize the genetic basis of VLCADD
- Review information and expand knowledge at each encounter
- Educational Resources:
Star-G
Fatty Acid Oxidation Support Group
References
Spierkerkoetter U, Sun B, Zytkovicz T, Wanders R, Strauss AW, Wendel U. (2003) MS/MS-Based newborn and family screening detects asymptomatic patients with very long chain acyl CoA dehydrogenase deficiency. J Pediatr. 143:335-342.
Goetzman ES, Wang Y, He M, Mohsen A-W, Ninness B, Vockely G. (2007) Expression and characterization of mutations in very long chain acyl-CoA dehydrogenase using a prokaryotic system. Mol. Genet. Metab. 91:138-147.
Gregersen N, Andresen BS, Corydon TJ, Corydon MJ, Olsen RK, Bolund L, Bross P. (2001) Mutation analysis in mitochondrial fatty acid oxidation defects: exemplified by acyl-CoA dehydrogenase deficiencies with special focus on genotype-phenotype relationship. Hum Mutat. 18:169-189.
Engbers HM, Dorland L, De Sain MGM, Eskes PF, Visser G. (2005) Rhabdomyolysis in early-onset very long chain acyl CoA dehydrogenase deficiency despite normal glucose after fasting. J. Inhert. Metab. Dis. 28:1151-1152.
Saudubray JM, Martin D, deLonlay P, Touati G, Poggi-Travert F, Bonnet D, Jouvet P, Boutron M, Slama A, Bianey-Saban C, Bonnefont JP, Tabier D, Kamoun P, Brivet MR. (1999) Recognition and management of fatty acid oxidation defets: A series of 106 patients. J Inher Metab Dis 22:488-502.
Cox GF, Souri M, Aoyama T, Rockenmacher S, Varvogli L, Rohr F, Hashimoto T, Korson MS. (1998) Reversal of severe hypertrophic cardiomyopathy and excellent neuropsychologic outcome in very-long-chain acyl-coenzyme A dehydrogenase deficiency. J Pediatr. 133:247-253.
Roe CR, Roe DS, Wallace M, Garritson B. (2007) Choice of oils for essential fat supplements can enhance production of abnormal metabolites in fat oxidation disorders Mol Genet Metab. 92:346-50.
Gillingham MB, Weleber RG, Neuringer M, Connor WE, Mills M, Van Calcar S, Ver Hooeve J, Wolff J, Harding CO. (2005) Effect of optimal dietary therapy upon visual function in children with long chain 3-hydroxy acyl CoA dehydrogenase and trifunctional protein deficiency. Mol Genet Metab. 86:124-133.
Roe CR, Sweetman L, Roe D, David F and Brunengraber H. (2002) Treatment of cardiomyopathy and rhabdomyolysis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. J Clin Invest. 110:259.
Spierkerkoetter U. (2007) Effects of a fat load and exercise on asymptomatic VLCAD deficiency. J Inher Metab Dis. 30:405.